Diagnostik 2.0: Next Generation Sequencing
als moderne diagnostische Erweiterung im medizinischen
B-Schutz am Beispiel von pockenartigen Läsionen bei einem Pferd
Rosina Ehmann a, Kristin Brandes b, Markus Antwerpen a, Mathias Walter a, Katja von Schlippenbach c, Elisabeth Stegmaier d, Sandra Eßbauer a, Joachim Bugert a, Jens Peter Teifke e, Hermann Meyer a
a Institut für Mikrobiologie der Bundeswehr, München
b Tierpathologie Augsburg, Deutschland
c Tierarztpraxis Zusamaltheim, Deutschland
d Makoa Farm, Tansania
e Friedrich-Loeffler-Institut, Greifswald – Insel Riems, Deutschland
Einleitung
Das Institut für Mikrobiologie der Bundeswehr (InstMikroBioBw) ist eine Forschungseinrichtung für den medizinischen B-Schutz mit breit gefächertem Spektrum. Ein zentraler Auftrag des Instituts ist es, Verfahren zur raschen und zweifelsfreien Diagnostik aller relevanten B-Agenzien zu etablieren und veränderten Anforderungen ständig anzupassen. Eine der größten Herausforderungen dabei ist die enorme und ständig wachsende Vielfalt an viralen und bakteriellen Erregern. So werden neue Arten und Subtypen entdeckt, während bereits bekannte Erreger sich in unterschiedlichen biologischen Nischen stetig verändern.
Seit wenigen Jahren bietet Next Generation Sequencing (NGS), ein Sammelbegriff für verschiedene Techniken der Hochdurchsatz-Sequenzierung, enorme Chancen zur breit angelegten Erregerdiagnostik. Allerdings ist NGS sowohl in Bezug auf technisches Know-how (labortechnisch, molekularbiologisch und bioinformatisch) als auch im Hinblick auf die Geräteausstattung eine herausfordernde Technik. Zur Implementierung müssen für verschiedene Probentypen (Serum, Stuhl, Nasenabstrich, Umweltprobe, …) zusätzlich jeweils individuelle Aufarbeitungsverfahren getestet und etabliert werden. Zur Evaluierung der Anwendungsmöglichkeiten von NGS in der Pockenvirusdiagnostik wurden im Rahmen dieser Arbeit beispielhaft Hautveränderungen eines Pferdes diagnostisch aufgearbeitet und ausgewertet. Die eingesetzten Methoden und Geräteplattformen wurden im Hinblick auf ihre Anwendbarkeit in der stationären und mobilen Diagnostik ausgewählt und verglichen. Der als ursächliches Agens identifizierte Erreger erwies sich als Vertreter einer neuen Virusspezies und wurde weitergehend charakterisiert.
Methoden
Als Probe dienten Hautbiopsien von knotigen, pockenähnlichen, ca. 0,5-1,5 cm großen Hautveränderungen eines Pferdes (Abbildung 1). Die Gewebeproben wurden zunächst mithilfe einer Schlagmatrix mechanisch zerkleinert und enzymatisch mit Proteinase K aufgeschlossen, um danach Nukleinsäure zu extrahieren (Abbildung 2). Die Nukleinsäureextraktion erfolgte zum einen maschinell in einem MagnaPure-Compact-System (Firma Roche®) sowie manuell mit der klassischen Phenol-Chloroform-Extraktions-Methode. Diese Methode lässt sich auch unter einfachen Laborbedingungen mit geringem technischem Aufwand durchführen und eignet sich gut für den mobilen Einsatz.
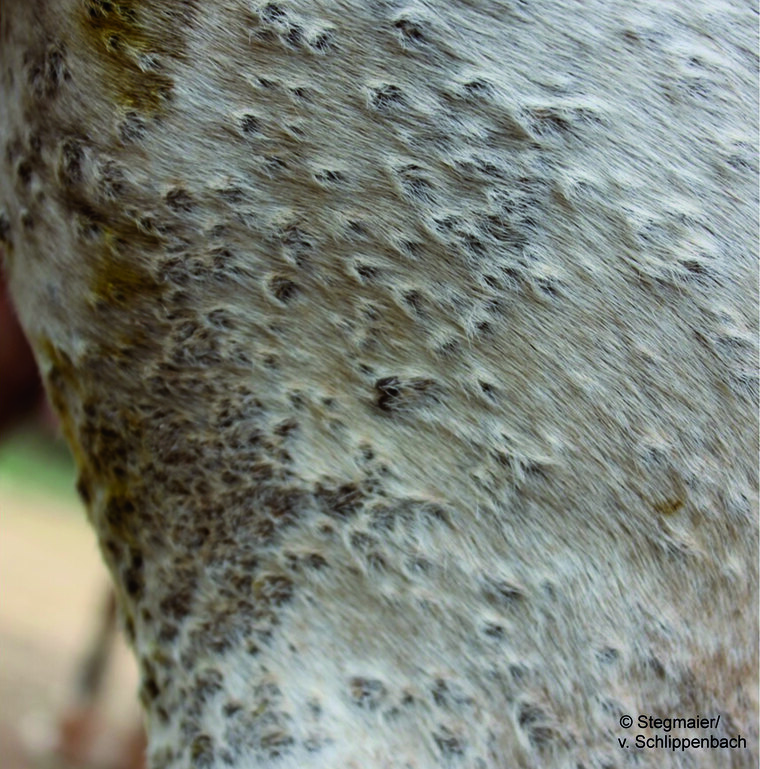
Abb. 1: Makroskopisches Erscheinungsbild der papulonodulären, ca. 0,5-1,5 cm großen Läsionen bei einem Pferd
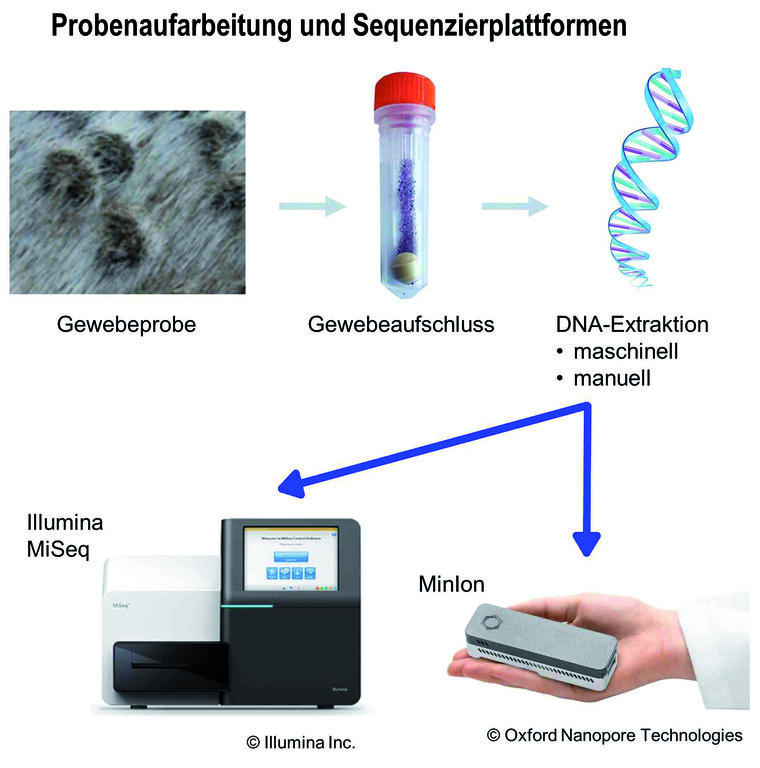
Abb. 2: Aufbereitung der Proben und Sequenzierung auf zwei verschiedenen Plattformen
Die Proben-DNA wurde anschließend auf zwei verschiedenen Sequenzierplattformen analysiert. Während die Illumina-MiSeq-Technologie (Firma Illumina®) eine exzellente Sequenziergenauigkeit und einen hohen Lesedurchsatz mit kurzen Fragmenten bietet, zeichnet sich die MinIon-Sequenzierplattform (Firma Oxford Nanopore Technologies®) durch eine größere Leseweite und ein handlicheres, auch im Feldbetrieb einsetzbares, Format aus.
Die gewonnen Rohdaten wurden bioinformatisch aufgearbeitet, zu einer Gesamtsequenz zusammengesetzt und anschließend durch vergleichende Analyse mit Datenbanksequenzen weitergehend ausgewertet. Die erhaltene virale Genomsequenz wurde annotiert, um die vorhandenen viralen Gene mit denen verwandter Viren zu vergleichen und in einen Kontext zu stellen.
Ergebnisse
Beide Sequenzierplattformen lieferten für die beschriebene Probe qualitativ hochwertige Rohdatensätze mit einer hohen Abdeckung der vorhandenen Nukleinsäuren. Die Art der DNA-Aufreinigung hatte nur einen geringen Einfluss auf die Sequenzierungsergebnisse, wobei die manuell aufgereinigte DNA der aus der automatisierten Extraktion leicht überlegen war.
Durch bioinformatische Aufarbeitung der Rohdaten konnte neben der Wirts-DNA vom Pferd genetische Information eines weiteren Organismus identifiziert werden, die im direkten Abgleich mit öffentlichen Genomdatenbanken (GenBank) Ähnlichkeit zu Molluscum contagiosum Virus (MOCV), einem beim Menschen weit verbreiteten Vertreter der Pockenviren, aufwies. Die einzelnen Sequenzfragmente dieses MOCV-ähnlichen Virus konnten wie Puzzlestücke zu einer Gesamtgenomssequenz von etwa 170 000 Basen zusammengesetzt werden.
Weitergehende phylogenetische Untersuchungen zur verwandtschaftlichen Beziehung des MOCV-ähnlichen Virus aus der Pferdehaut-Biopsie zeigten, dass das untersuchte Virus charakteristisch abweichende Genommerkmale besitzt, die es von den bisher bekannten MOCV-Stämmen unterscheidet und im phylogenetischen Baum auf einen dem MOCV nahe verwandten, aber eigenen Zweig positionieren (Abbildung 3). Diese Daten legen nahe, dass das identifizierte Virus einer neuen, bisher nicht beschriebenen Virusspezies angehört, die vorläufig equines Molluscum-contagiosum-ähnliches Virus bezeichnet wurde (EMCLV für engl. „equine molluscum contagiosum-like virus“).
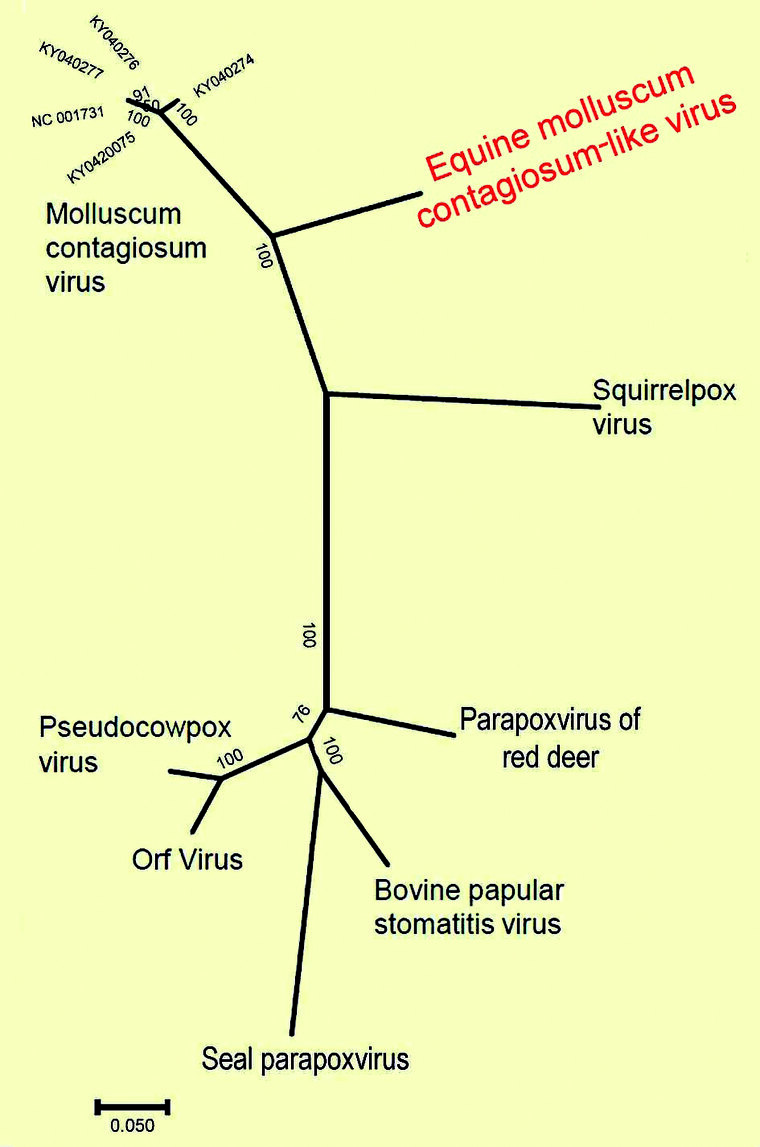
Abb. 3: Phylogenetische Analyse des mittels NGS identifizierten neuen Virus mit Positionierung auf einem von MOCV und anderen Pockenspezies klar abgegrenzten Zweig
Durch Kombination der jeweiligen Vorteile beider Sequenzierplattformen – hohe Genauigkeit durch den Illumina-Datensatz bei gleichzeitig genauer Auflösung komplexer, repetitiver Genomabschnitte durch die längeren MinIon-Sequenzinformationen – gelang es, eine zuverlässige Gesamtgenomsequenz für das neue Virus zu erstellen. Nach Identifizierung und Annotation der Gene in EMCLV erfolgte ein Vergleich mit dem Genrepertoire von MCV. Dabei konnten mehrere Beobachtungen gemacht werden.
EMCLV kodiert 160 Gene. Im Vergleich zu MOCV fehlen dem equinen Virus 24 Gene, wobei es über 21 eigene Gene verfügt, die nicht in MOCV vorkommen. Gleichzeitig weist das Genom deutliche Anpassungen an Equiden als Wirtstiere auf.
Pockenviren haben die Fähigkeit, Gene vom Wirt zu übernehmen und in ihr Genom aufzunehmen. Die verwandtschaftliche Herkunft solcher wirtsassoziierter Gene in Pockengenomen – EMCLV besitzt vier Gene, die vom Pferd stammen – sind eindeutige Anpassungsmerkmale an bestimmte Spezies. Zusätzlich konnten im Genom von EMCLV zwei bei Viren bisher nicht festgestellte Gene identifiziert werden, die Proteine des Immunsystems des Wirtstieres imitieren und somit zwei neue Strategien darstellen, wie Viren die Immunantwort des Wirts modulieren können.
Diskussion und Schlussfolgerung
NGS ist ein vielseitig einsetzbares Werkzeug in der modernen Diagnostik, benötigt aber neben den technischen Voraussetzungen auch ein detailliertes Erarbeiten probenspezifischer Aufarbeitungsverfahren. In der Pockenvirusdiagnostik ist die Verwendung „echter“ Proben für viele Erreger aufgrund der Sicherheitsbestimmungen und/oder Verfügbarkeiten (Affenpockenvirus und Variolavirus) stark eingeschränkt, sodass in diesem Projekt zur Verwendung von authentischem Testmaterial auf eine tierische Probe zurückgegriffen wurde.
Die vorgestellte Arbeit konnte zeigen, dass Next Generation Sequencing als Werkzeug für die diagnostische Aufarbeitung von Gewebeproben mit Verdacht auf durch Pockenviren bedingte Erkrankungen eine exzellente Erweiterung bereits etablierter Verfahren – wie z. B. der PCR – ist. Zwei unterschiedliche Sequenzierplattformen konnten dabei unabhängig voneinander den ursächlichen Erreger erfolgreich detektieren. Dies eröffnet neue Möglichkeiten sowohl für die stationäre als auch für die mobile Diagnostik im Repertoire der Bundeswehr.
Neben den technisch-methodischen Erkenntnissen zur Diagnostik konnten unerwartet neue Einblicke in die Pockenvirusbiologie durch die Entdeckung einer neuen Virusspezies, dem equinen Molluscum-contagiosum-ähnlichen Virus erzielt werden. Die Identifikation von zwei bisher bei Viren nicht bekannten Genen, die im Zusammenhang mit der Modulation des Wirts-Immunssystems stehen, eröffnen interessante Fragestellungen für die Zukunft.
Die vorgestellte Arbeit zeigt, dass wehrmedizinische Forschung in der Bundeswehr als „Nebenprodukt“ disziplinenübergreifend die Grundlagenforschung voranbringen und neue Impulse für die zivile Forschung initiieren kann.
Literaturhinweise
- Günther T, Haas L, Alawi M, Wohlsein P, Marks J, Grundhoff A, Becher P, Fischer N: Recovery of the first full-length genome sequence of a parapoxvirus directly from a clinical sample. Sci Rep 2017; 7: 1–8. mehr lesen
- Huang B, Jennsion A, Whiley D, McMahon J, Hewitson G, Graham R, Jong AD, Warrilow D: Illumina sequencing of clinical samples for virus detection in a public health laboratory. Sci Rep 2019; 9: 1–8. mehr lesen
- López-Bueno A, Parras-Moltó M, López-Barrantes O, Belda S, Alejo A: Recombination events and variability among full-length genomes of co-circulating molluscum contagiosum virus subtypes 1 and 2. J. Gen. Virol. 2017; 98, 1073–1079. mehr lesen
- Minogue TD, Koehler JW, Stefan CP, Conrad TA: Next-Generation Sequencing for Biodefense: Biothreat Detection, Forensics, and the Clinic. Clin. Chem. 2019; 65: 383–392. mehr lesen
- Rahaley RS, Mueller RE: Molluscum contagiosum in a horse. Vet. Pathol. 1983; 20: 247–250. mehr lesen
Manuskriptdaten
Zitierweise
Ehmann R, Brandes K, Antwerpen M, Walter M, v. Schlippenbach K, Stegmaier E, Essbauer S, Bugert J, Teifke JP, Meyer H: Diagnostik 2.0: Next Generation Sequencing als moderne diagnostische Erweiterung im medizinischen B-Schutz am Beispiel von pockenartigen Läsionen bei einem Pferd (Vortrags-Abstract). WMM 2019; 63(12): 406-408.
Für die Verfasser
Stabsveterinär Rosina Ehmann
Institut für Mikrobiologie der Bundeswehr
Neuherbergstr. 11, 80937 München
E-Mail: rosinaehmann@bundeswehr.org
Der Beitrag wurde als Vortrag im Rahmen des Wettbewerbs um den Heinz-Gerngroß-Förderpreis beim 50. Jahreskongress der Deutschen Gesellschaft für Wehrmedizin und Wehrpharmazie e. V. in Leipzig am 11. Oktober 2019 mit dem 2. Preis ausgezeichnet.