ONKOLOGIE: KASUISTIKEN
Das diffus großzellige B-Zell-Lymphom der Hypophyse: Fallbericht einer seltenen Manifestation und Einordnung in die Literatur
Diffuse large B-cell lymphoma of the pituitary gland: Case report of a rare disease and review of literature
Conn Rothera, Niklas Gebauerb, Thomas Mayera, Armin Rieckea, Matthias Müllera, Carsten Hackenbrochc,
Chris Schulzd, Konrad Steinestele, Hanno M. Wittea,b,1
a Bundeswehrkrankenhaus Ulm, Klinik für Innere Medizin/Hämatologie und Onkologie
b Universitätsklinikum Schleswig-Holstein, Campus Lübeck, Klinik für Hämatologie und Onkologie
c Bundeswehrkrankenhaus Ulm, Klinik für diagnoistische und interventionelle Radilogie und Neuroradiologie
d Bundeswehrkrankenhaus Ulm, Klinik für Neurochirurgie
e Bundeswehrkrankenhaus Ulm, Institut für Pathologie und Molekularpathologie
Zusammenfassung
Wir präsentieren die seltene Manifestation eines diffus großzelligen B-Zell-Lymphoms (DLBCL) im vorderen Anteil der Hypophyse. Bei initialer Präsentation bestand ein partieller Hypopituarismus und eine inkomplette Okulomotoriusparese. Unter dem Verdacht eines Hypophysenmakroadenoms wurde eine transsphenoidale Resektion durchgeführt. Die anschließende histopathologische Begutachtung brachte das Ergebnis eines malignen Lymphoms hervor. Die Immunchemotherapie mittels R-CHOP (Rituximab, Cyclophosphamid, Doxrubicin, Vincristin, Prednisolon) führte zu einer kompletten Remission.
Die Analyse der vorliegenden Literatur konnte insgesamt 36 Fälle von Non-Hodgkin-Lymphomen der Hypophyse identifizieren. Der Nicht-Keimzentrums(non-GCB oder ABC)-Typ eines DLBCL gemäß der Hans-Klassifikation kommt etwas häufiger vor als der Keimzentrums-GCB-Typ. Eine visuelle Störung, hervorgerufen durch eine Kompression des Chiasma opticum oder des Nervus opticus, oder endokrinologische Störungen der Adenohypophyse, mit oder ohne Auftreten eines Diabetes insipidus, gehören zu den am häufigsten dokumentierten Symptomen bei diesen Patienten.
Stichworte: Diffus großzelliges B-Zell-Lymphom, Hypophyse, Prognose, Immunchemotherapie
Summary
We present a rare case of diffuse-large-B-cell lymphoma (DLBCL) occurring in the anterior pituitary lobe. Initial clinical presentation included partial hypopituarism and incomplete oculomotor palsy. Transsphenoidal resection was performed on suspicious diagnosis of macroadenoma. Subsequent histopathological assessment revealed malignant lymphoma. Immunochemotherapy with R-CHOP achieved complete remission.
Review of literature detected 36 cases of non-Hodgkin-lymphoma of the pituitary gland, most commonly DLBCL with a slight emphasis on the non-GCB (ABC)-type according to the Hans classification. Visual aberration due to compression of the optic chiasm or the optic nerve and also endocrine insufficiency of the anterior lobe with or without developing Diabetes insipidus were the most frequently reported symptoms in those patients.
Keywords: diffuse-large-B-cell lymphoma, pituitary gland, prognosis, immunochemotherapy
Einleitung
Das diffus großzellige B-Zell-Lymphom, not otherwise specified, (DLBCL, NOS) ist mit 25-35 % der häufigste Subtyp der aggressiven Non-Hodgkin-Lymphome (NHL). Die Inzidenz steigt mit zunehmendem Lebensalter (7/100 000 pro Jahr). Männer erkranken häufiger als Frauen. Neben der für Lymphome typischen Lymphadenopathie kommt es bei DLBCL in 40 % der Fälle zu extranodalen Manifestationen. In 10-25 % der Fälle liegt eine Knochenmarkinfiltration vor. Prinzipiell kann das DLBCL jedes Organ befallen. Zu den typischen Extranodalmanifestationen gehören Gastrointestinaltrakt, Knochen, Speicheldrüsen, Schilddrüse, Niere, Nebennieren, Leber und ein Befall der Hoden [12][43][44]. Das primäre ZNS-Lymphom (PZNSL) wird in der gültigen WHO-Klassifikation aus 2016 als eigenständige Entität aufgeführt, bei dem das mediane Erkrankungsalter bei 61 Jahren liegt. Der einzige identifizierte Risikofaktor ist eine Immunsuppression (z. B. HIV-Infektion). Es kann neben dem Großhirn auch das Rückenmark, die Augen oder den Liquor cerebrospinalis befallen [10][39][44].
Aus biologischer Sicht weisen beide Entitäten Ähnlichkeiten auf. Immunhistochemisch lassen sich für beide Entitäten die typischen B-Zell-Marker CD19, CD20, CD22, CD79a und PAX5 nachweisen. Die Expression von CD5 geht mit einer schlechten Prognose einher. Gemäß der Cell-of-origin (COO) Klassifikation nach HANS et al. werden die immunhistochemischen Marker CD10, BCL6 und MUM1/IRF4 berücksichtigt. Ein Ursprung in lymphofollikulären Keimzentrumszellen (GCB-Typ) liegt demnach in 56 % der Fälle, der nicht-Keimzentrumstyp, welcher sich durch aktivierte B-Zellen auszeichnet, liegt in 32 % der Fälle vor (non-GCB oder ABC-Typ). 11 % der Fälle lassen sich entsprechend des Hans-Klassifikators nicht klassifizieren [12]. Die Angabe der Expression von BCL2 variiert in der Literatur zwischen 47-84 %. Die gleichzeitige Expression von MYC und BCL2 entspricht dem Vorliegen eines ,,double-expressor lymphoma (DEL)‘‘, welches ebenfalls mit einer verschlechterten Prognose assoziiert ist. Der Ki-67 liegt bei mindestens >40 % und oftmals >90 %. CD10 wird im PZNSL in <10 % auf der Zelloberfläche exprimiert, während sich BCL6 (60-80 %) und MUM1/IRF4 (90 %) in den meisten Fällen nachweisen lassen [44].
Zytogenetisch wird standardmäßig eine Translokation des c-MYC-Gens mittels FISH-Diagnostik (Fluoreszenz-in-situ-Hybridisierung) überprüft. Bei gleichzeitigem Rearrangement im BCL-2- und /- oder BCL-6-Gen in der Anschlussanalyse liegt ein sogenanntes ,,Double-Hit‘‘ bzw. ,,Triple-Hit‘‘ Lymphom (DHL/THL) vor. Jüngste Ergebnisse von ROSENWALD et al. suggerieren, dass für das c -MYC-Rearrangement entscheidend ist, ob es sich um ein Immunglobulin-Gen als Translokationspartner handelt. Die Prognose dieser High-Grade Lymphome ist ungünstig [36][44].
Die Standardchemotherapie für das primäre ZNS-Lymphom besteht aus einer Methotrexat beinhaltenden Induktionstherapie mit anschließender Hochdosischemotherapie und autologer Stammzelltransplantation (z. B. das MATRix-Protokoll der IELSG322 ; Methtotrexat, Cytarabin, Thiotepa, Rituximab), während die standardmäßige Behandlung des DLBCL, NOS eine weitaus weniger intensive alleinige R-CHOP-ähnliche Immunchemotherapie (Rituximab, Cylcophosphamid, Doxorubicin (Hydroxydaunorubicin), Vincristin (Oncovin), Prednison) umfasst [8][16].
Mit dieser Therapie wird für das DLBCL, NOS wird eine allgemeine Ansprechrate (ORR) von 90 % erreicht. Die Heilungsrate liegt bei 60-70 % [16].
Eine extranodale Manifestation im Bereich der Hypophyse lässt sich nur in wenigen Fällen nachweisen. Diese Patienten fallen oftmals durch endokrinologisch assoziierte Symptome oder eine bitemporale Hemianopsie, hervorgerufen durch ein verdrängendes supraselläres Wachstum, auf. Die Lebenszeitprävalenz eines Hypophysentumors, unabhängig von dessen Entität, liegt bei 16,7 % [7][29]. Therapeutisch sind neben einer transsphenoidalen Resektion auch strahlentherapeutische Verfahren bei Inoperabilität etabliert. Das Prolaktinom beispielsweise lässt sich medikamentös behandeln [11][14][23]. Lymphom-Befälle der Hypophysenregion wurden bisher lediglich im Rahmen von Fallpräsentationen beschrieben [1-3][5][6][13][17-22][24-28][30-35][37][38][40-42][45-48]. Eine explizite Zuordnung zur Entität des DLBCL, NOS oder dem primären ZNS-Lymphom mit den daraus resultierenden therapeutischen Konsequenzen ist bisher nicht erfolgt.
In vorliegendem Fall präsentierte sich ein initial transsphenoidal reseziertes Inzidentalom als (para-)selläre Manifestation eines DLBCL. Mittels Immunchemotherapie konnte nach 6 Zyklen R-CHOP21 eine komplette Remission erreicht werden. Eine besondere Schwierigkeit stellte die Einordnung des Malignoms in die gültige WHO-Klassifikation (DLBCL, NOS versus PZNSL) dar.
Fallbericht
Die initiale auswärtige Vorstellung der 55-jährigen Patientin erfolgte im August 2018 bei Diplopie, Anisokorie, rechtsseitiger Ptosis und Kopfschmerzen. In der klinischen Untersuchung fand sich eine partielle rechtsseitige Okulomotoriusparese mit Stauungspapille. Es konnte ein basales TSH von 0,001 μU/l (Norm 0,2-3,4 μU/l) bei normwertigen fT4 und fT3, normwertiges basales Cortisol und Prolaktin gemessen werden.
Die Schädel-MRT (Magnetresonanztomografie) zeigte eine 1,5x1,5x3,5 cm messende selläre Raumforderung mit beidseitigem Einbruch in den Sinus cavernosus und Ummauerung der Arteria carotis interna.
Im Verlauf von zwei Monaten entwickelte sich eine zunehmende Fatique-Symptomatik. Laborchemisch trat eine Prolaktinämie von 1372 μU/ml (Normwert ≤ 49 μU/ml), im CRH-Test ein erniedrigtes ACTH und ein Basal-Cortisol mit erniedrigtem Inkrement auf, sodass bei Insuffizienz der corticotropen, gonadotropen (in postmenopausaler Situation) und beginnend thyreotropen Achse, gleichzeitig normwertiger somatotroper Achse und Enthemmungshyperprolaktinämie, eine Hydrocortisonsubstituion begonnen wurde. Zudem bestand eine Hyponatriämie. Bei fehlendem Anhalt auf einen Diabetes insipidus wurde ein partieller Hypopituarismus festgestellt. Der Liquor cerebrospinalis stellte sich als unauffällig dar.
Unter der Verdachtsdiagnose eines hormoninaktiven Makroadenoms oder eines Meningeoms wurde eine operative Resektion geplant. In der Planungs-CT (Computertomografie) zeigte sich die dem Tuberculum sellae mittlerweile flächig anliegende Raumforderung progredient mit konsekutiver ossärer Arrosion des Dorsum sellae und des rechten Processus clinoideus.
Im Dezember 2018 erfolgte die transsphenoidale Tumorexstirpation. Der postoperative Verlauf stellte sich ohne Auftreten eines Diabetes insipidus komplikationsarm dar. Histopathologisch konnte eine Infiltration der Hypophyse durch ein B-Zell-NHL festgestellt werden.
Histopathologie
Die Histopathologie zeigte eine diffuse Hypophyseninfiltration durch blastäre lymphatische Zentro- und Immunoblasten mit deutlich erhöhter Aktivität ohne Beteiligung der Pars intermedia oder der Neurohypophyse. Immunhistochemisch lag der Ki-67-Index bei 80 %. Die B-Zellen exprimierten CD20, BCL-2, mehrheitlich BCL-6 sowie MUM1/IRF4 zu 20 % bei immunhistochemischer Negativität für CD10. In der Zusammenschau ließ sich der Befund daher mit einem DLBCL vom Non-GCB-Typ nach dem Hans-Algorithmus vereinbaren [12]. Es konnte keine c-MYC-Translokation nachgewiesen werden.
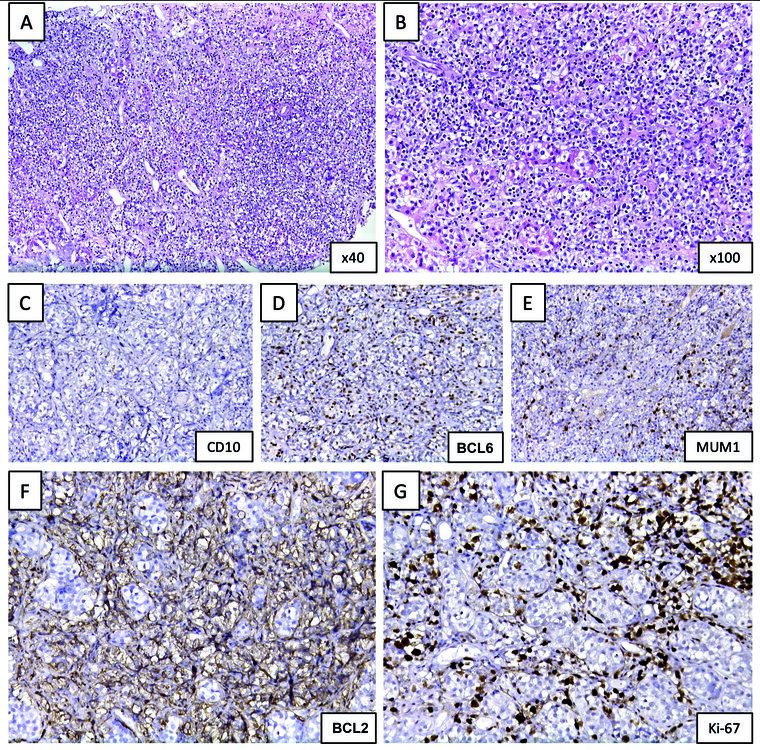
Abb. 1: Histopathologische und immunhistochemische Diagnostik aus dem Hypophysenresektat
(A) und (B): H.E.-gefärbte Schnittpräparate der infiltrierten Hypophyse
(C): Es besteht kaum Färbereaktivität mit dem Antikörper gegen CD10.
(D): Der überwiegende Anteil der infiltrierenden Blasten positiv für BCL-6.
(E): MUM1 wird in ca. 20 % der Blasten exprimiert.
(F) und (G): Die Blasten sind BCL-2-positiv und zeigen eine rasante Proliferation mit dem Marker Ki-67.
Klinischer Verlauf
Im Februar 2019 wurde uns die Patientin zum Restaging und Einleitung einer zytoreduktiven Therapie nach dem MATRix-Protokoll überwiesen. In der klinischen Untersuchung präsentierte sie sich im ECOG-Performancestatus 1 und residuell verbliebender Okulomotoriusparese. Die Post-Resektions-Schädel-MRT Aufnahmen zeigten einen Progress der Lymphommanifestation mit 2,3x0,8x2,2 cm im Bett der exstirpierten Adenohypophyse und den äußeren Wand- und luminalen Anteilen des linken Sinus sphenoidales (Abbildung 2 a-d) ohne weitere zentrale Manifestationen. Eine extrakranielle Manifestation konnte in einer PET-CT (Positronenemissionstomografie) nicht dargestellt werden. Für einen Knochenmarkbefall ergaben sich sowohl in der PET-CT als auch in der Knochenmarksbiopsie keine Hinweise.
Während eine initiale referenzpathologische Begutachtung zunächst das Vorliegen eines PZNSL favorisierte, erfolgte im Verlauf die Zuordnung hin zu einem DLBCL, NOS aufgrund der anatomischen Lage sowie der fehlenden Zeichen einer zentralen Infiltration. Diese Argumentation stützend diente die Beschreibung, dass die Adenohypophyse, im Gegensatz zur Neurohypophyse, aus anatomischer Sicht kein Teil des ZNS darstellt. Die Kasuistik wurde wiederholt im interdisziplinären Lymphomboard diskutiert. Entsprechend der dann festgelegten Diagnose eines DLBCL (Non-GCB Typ) im Stadium IVAE nach Ann-Arbor (CNS-IPI 3 0) entschieden wir uns für eine R-CHOP basierte Therapie.
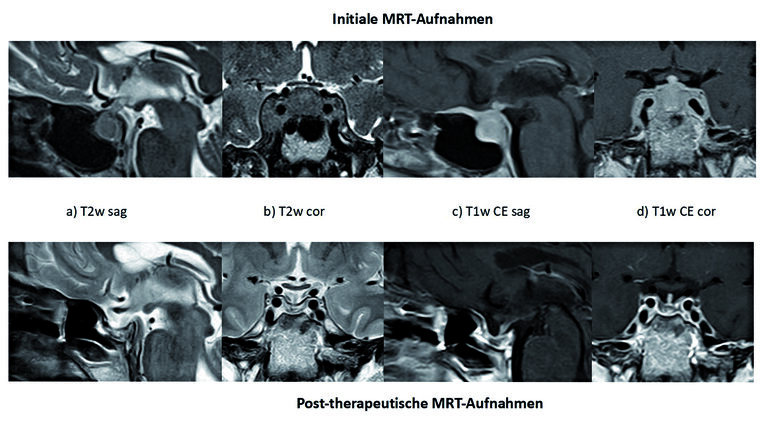
Abb. 2: MRT-Aufnahmen der Sellaregion prä und posttherapeutisch bei DLBCL, NOS der Hypophyse
Obere Reihe – initiales MRT, untere Reihe – post- therapeutisches MRT.
Von links nach rechts: a) T2 sag, b) T2 cor, c) T1 CE, d) T1 CE
Das initiale MRT zeigt eine rundliche in T2w homogene und relativ hypointense Auftreibung der Hypophyse mit Beteiligung des Hypophysenstiels. Gut abgrenzbar die Ausdehnung in den Sinus cavernosus beidseits. Nach Kontrastmittelgabe zeigt sich eine homogene Kontrastmittelanreicherung der Raumforderung mit duraler Ausdehnung nach ventral und dorsal.
Post-therapeutisch findet sich wieder eine normalgroße Hypophyse mit schlankem Hypohysenstiel sowie eine regelrechte Darstellung des beidseitigen Sinus cavernosus. Nach Kontrastmittelgabe zeigt sich eine fehlende zentrale Kontrastierung der Hypohyse, wie bei einer Nekrose.
Therapie
Bis Juni 2019 wurden insgesamt 6 Zyklen R-CHOP-21 appliziert, wodurch es zu einer vollständigen Regredienz der Hirnnervenausfälle kam und ein Auftreten maßgeblicher adverser Therapiereaktionen ausblieb. In Interims- und posttherapeutischen Schädel-MRT-Aufnahmen zeigte sich eine sehr gute partielle Remission (VGPR) und nach Therapieabschluss eine komplette Remission (CR) (Abbildung 2 a-d). In den dazu konkordant durchgeführten PET-MRT Aufnahmen war keine suspekte Avidität detektierbar. Zu Beginn des Nachbeobachtungsintervalls präsentierte sich die Patientin ohne fokale neurologische Defizite unter unverändert bestehender Notwendigkeit zur Cortisolsubstitution.
Einordnung in die Literatur
TARABAY et al. [46] konnten in ihrer Literaturdurchsicht bis 2015 insgesamt 33 Fälle eines NHL der Hypophyse detektieren. Die anschließende Recherche im Rahmen dieser Arbeit konnte ab 2015 bis einschließlich November 2019 drei weitere Fälle eines Befalls der Hypophyse durch ein NHL herausarbeiten. In der Online-Version des Beitrags steht eine tabellarische Übersicht aller bisher publizierten Fälle (n=36) zur Verfügung.
Einschließlich des hier präsentierten Falls lag in 83 % (n = 31) ein B-NHL vor. Der Subtyp eines DLBCL lag in 70 % (n = 26), ein MALT-Lymphom (Mukosa-assoziiertes lymphatisches Gewebe) in 5 % (n = 2) und ein Burkitt-Lymphom in einem Fall (3 %) vor. In 5 Fällen lag ein peripheres T-Zell-Lymphom und in einem Fall ein NK-Zell Lymphom vor. Fünf Fälle (13 %) wurden vor der Erstformulierung der heutig gültigen WHO-Klassifikation publiziert, sodass keine adäquate Subspezifizierung vorgenommen werden konnte.
In der Subgruppe der DLBCL waren 77 % (n = 21) primär in der Hypophyse auftretend (PPL). Es liegt eine leichte Prädominanz des weiblichen Geschlechtes mit 63 % (n = 17) vor. Das Auftreten der Fälle wird im Alter von 37 bis 86 Jahren (im Median 64 Jahre) beschrieben.
Eine teilweise oder vollständige Hypophyseninsuffizienz trat in 85 % und eine Beteiligung der Neurohypophyse in Form einer Manifestation eines Diabetes insipidus centralis in 27 % der dokumentierten Fallberichte auf. Kopfschmerzen wurden in 48 % und das Auftreten einer B-Symptomatik in 30 % der Fälle beschrieben. Weitere häufige Symptome waren Sehstörungen und Hirnnervenausfälle, meist eine bitemporale Hemianopsie oder eine Okulomotoriusparese. Seltener war eine Abduzensparese oder die Affektion eines Trigeminusastes vorliegend.
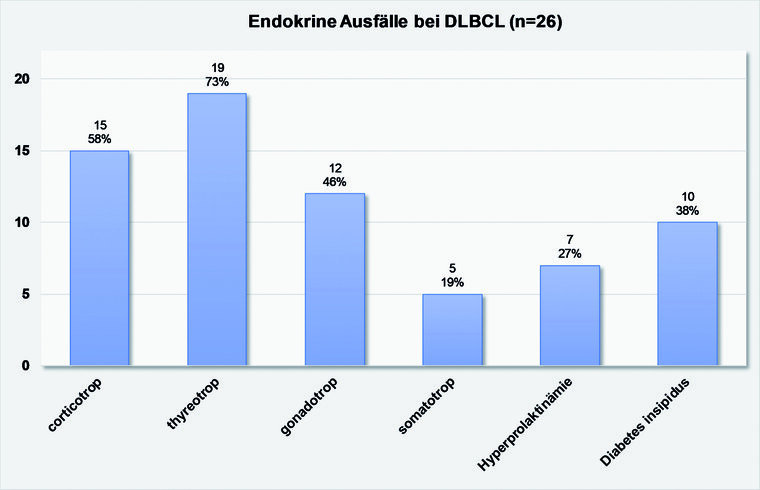
Abb. 3: Endokrine Ausfälle bei Patienten mit DLBLC der Hypophyse (N = 26)
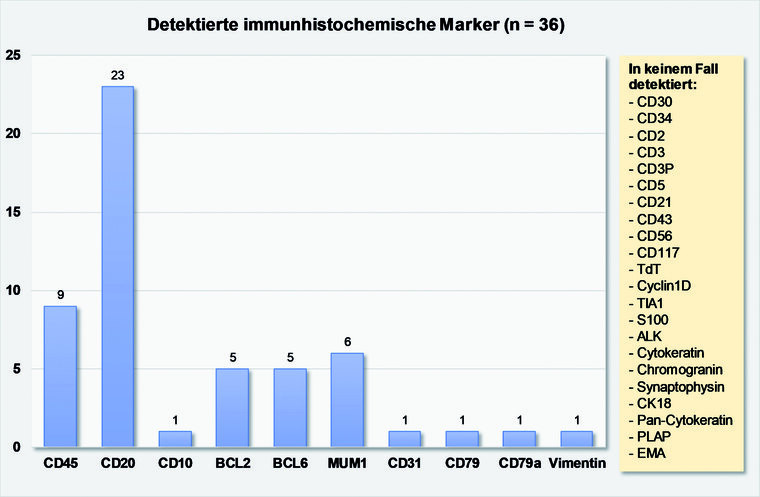
Abb. 4: Grafische Darstellung der detektierten immunhistochemischen Marker anhand der bisher publizierten Falldarstellungen (n = 36)
Kernspintomografisch kam bei 88 % der Patienten eine selläre Raumforderung oder eine Vergrößerung der Hypophyse zur Darstellung. Eine zusätzliche Kompression des Infundibulums oder eine supraselläre Raumforderung bestand in 38 % der MRT-Aufnahmen. Eine isolierte Manifestation des Infundibulums oder suprasellär kamen deutlich seltener vor (11 %). Eine Ausbreitung in den Sinus cavernosus oder sphenoidales war in 42 %, eine Kompression des Chiasma opticum in 43 % zu detektieren. Eine Infiltration per contuinitatem in Strukturen des zentralen Nervensystems (ZNS), vor allem des Hypothalamus, wurde in 23 % der Fallberichte beschrieben.
In zwei der berichteten Fälle wurde die seltene Subform eines intravaskulären DLBCL beschrieben. Der immunhistochemischen Klassifikation nach dem Hans-Algorithmus folgend, wurde der Non-GCB-(ABC-Typ mit 27 % im Vergleich mit dem GCB-Typ mit 11 % häufiger zugeordnet. Allerdings lag in 61 % der Fälle keine Einordnung nach dieser Klassifikation vor. Die immunhistochemische Konstellation der in der Literatur publizierten Fallberichte ist in Abbildung 3dargestellt. Zwei Falldokumentationen war keine immunhistochemische Diagnostik zu entnehmen. Zytogenetische Daten sind lediglich einer publizierten Kasuistik von MOSHKIN et al. aus 2009 zu entnehmen [31]. Hier konnte mittels FISH-Diagnostik sowohl eine BCL-2 und eine BCL -6 Translokation beschrieben werden.
Diskussion
Ein Non-Hodgkin-Lymphom der B-Zell-Reihe mit primärer Manifestation oder einer Mitbeteiligung der Hypophyse ist eine Rarität. Nach wie vor unklar ist die eindeutige diagnostische Zuordnung zu einem DLBCL, NOS oder einem PZNSL. Sowohl für das DLBCL, NOS als auch für das PZNSL ist ein häufigeres Auftreten für das männliche Geschlecht, beschrieben mit einer Ratio von 3:2. Interessanterweise manifestiert sich das B-Zell-NHL der Hypophyse in der bisher vorliegenden Literatur häufiger beim weiblichen Geschlecht, mit einer Ratio von ebenfalls etwa 3:2.
Therapeutische Optionen und Überlegungen
In der Regel wird unter dem Verdacht eines Hypophysenmakroadenoms eine transsphenoidale Resektion indiziert. Sobald die histopathologische Begutachtung die Diagnose eines B-Zell-NHL hervorbringt, wird in den meisten Fällen eine Immunchemotherapie oder eine Induktionschemotherapie mit Hochdosis-Chemotherapie und anschließender autologer Stammzelltransplantation eingeleitet. Die Rolle einer involved field Radiotherapie (IF) kann bei unzureichenden Nachbeobachtungsdaten nicht abschließend bewertet werden. Bislang konnte für eine adjuvante Radiatio-haltige Therapie gegenüber einer alleinigen (Immuno-) Chemotherapie kein verbessertes Überleben gezeigt werden [46].
Eine initiale Radiatio einer Nicht-Bulky-Läsion außerhalb einer rein symptomatischen Indikation ist bisher nicht hinreichend evaluiert [4][15]. Bei PET-positiven Restmanifestationen hingegen verbessert die Radiatio das Überleben von DLBCL-Patienten signifikant [9]. Dies kann jedoch anhand der vorliegenden Daten nicht abschließend bewertet werden.
Insbesondere ist zu betonen, dass sowohl in Protokollen für das DLBCL als auch das PZNSL der monoklonale CD20-Antikörper Rituximab fester Bestandteil der Standardbehandlung ist. Immunhistochemisch weisen 88,5 % der primär an der Hypophyse manifestierten DLBCL eine Expression von CD20 auf. In 7,9 % der Fälle liegt keine immunhistochemische Diagnostik vor. Der Einsatz einer Rituximab beinhaltenden Immunchemotherapie wurde in nur 47 % der hier evaluierten Fälle berichtet. Das CHOP-Protokoll kam in 23 % (n = 6) der Fälle zur Anwendung. Das mediane Gesamtüberleben (OS) beträgt 12 Monate und das progressionsfreie Überleben (PFS) 10,5 Monate.
Der Stellenwert von Methtrexat
Das ZNS-gängige Methotrexat besitzt sowohl in der Behandlung des PZNSL als auch in der Therapie des DLBCL, NOS mit hohem CNS-IPI und damit einhergehendem hohen Risiko einer zentralen Manifestation einen elementaren Stellenwert. Der Stellenwert begründet sich auf der prophylaktischen Wirkung hinsichtlich eines zentralen Befalls. In der vorliegenden Kasuistik lag der CNS-IPI bei 0. Daher bestand, bei nicht eindeutigem Nachweis einer Lymphom-assoziierten ZNS-Läsion, keine Indikation zur therapeutischen Hinzunahme von Methotrexat. In der Vielzahl der in der Literatur aufgeführten Fälle wurde eine direkte ZNS-Infiltration oder die Kompression zentraler Strukturen, wie dem Chiasma Optimum oder dem Infundibulum beschrieben. Bei teilweise kurzen oder fehlenden Nachbeobachtungen kann der Stellenwert einer methotrexathaltigen ZNS-Prophylaxe nicht sicher beurteilt werden. Die intrathekale Applikation von Chemotherapeutika besitzt keinen evidenzbasierten Stellenwert für die ZNS-Prophylaxe.
Das Problem der eindeutigen klinischen und hämatopathologischen Zuordnung
Die Mehrzahl der Autoren ordnet das primär hypophysär manifestierte B-Zell-NHL eher den PZNSL zu. Dies erscheint, anatomischen Überlegungen folgend, nicht plausibel. Embryogenetisch gliedert sich die Adenohypophyse aus der Rathke-Tasche an das ZNS an. Dem Kapillarendothel fehlen die für die Blut-Hirn-Schranke typischen „tight junctions“.
In der hier dargestellten Kasuistik konnte aus histopathologischer Sicht keine eindeutige zentrale Manifestation beschrieben werden, sodass die initiale Diagnose PZNSL revidiert wurde. Daraufhin wurde eine für das DLBCL, NOS typische Immunchemotherapie nach dem R-CHOP Schema initiiert. Der Therapieerfolg wurde mittels Interims- und Abschluss-PET-MRT evaluiert. Aus der oben aufgeführten Argumentation hervorgehend, wurde von einer methotrexathaltigen ZNS-Prophylaxe abgesehen. Auch ein Therapiewechsel auf ein Hochdosis-Chemotherapie-Konzept erschien vor dem Hintergrund nicht sinnvoll. Diese differenzialdiagnostischen Überlegungen und die gezielte Entscheidung zu einer sinnvollen, aber weniger intensiven, zytoreduktiven Therapie geht aus keiner der bisher publizierten Falldarstellungen hervor.
Während Hypophysenadenome klinisch häufig inapparent verlaufen bzw. mit isolierten Sehstörungen oder, bei endokrin aktiven Tumoren am häufigsten, mit einer Hyperprolaktinämie einhergehen, imponiert die Manifestation eines B-Zell-NHL meist mit dem Ausfall mehrerer endokrinologischer Achsen des Hypophysenvorderlappens. Seltener stellt sich ein Diabetes insipidus ein. Kompressionsbedingte Sehstörungen gehören zu den häufiger beschriebenen Erstsymptomen von hypophysären B-Zell-NHL. Eine schnell größenprogrediente Raumforderung und der rasch auftretende Ausfall endokriner Achsen bis hin zum Vorliegen einer Hypophysenvorderlappeninsuffizienz sollten differentialdiagnostisch an maligne Veränderungen denken lassen.
Therapeutische Konsequenz
Die zentrale Frage, die sich für das hier präsentierte Krankheitsbild formulieren lässt, zielt auf die endgültige Zuordnung des an der Hypophyse manifestierten B-Zell-NHL ab. Aus der abschließenden Klärung dieser Fragestellung leitet sich das optimale therapeutische Konzept für diese Entität ab. Im Falle einer Zuordnung zu einem PZNSL würde das Anstreben einer Hochdosis-Chemotherapie mit anschließender autologer Stammzelltransplantation das Procedere der Wahl sein. Sollte sich stets eine eindeutige Zuordnung zum DLBCL, NOS ergeben, wird eine R-CHOP-haltige Immunchemotherapie als Strategie der Wahl favorisiert werden. Die Rolle der Radiotherapie muss, wie bereits oben erwähnt, zusätzlich diskutiert werden. Die Bestrahlungstherapie von PET-aviden Restmanifestationen ist mit großer Wahrscheinlichkeit eine Option, die das Therapieergebnis langfristig verbessern kann. Gegebenenfalls lassen sich B-Zell-NHL, die sich primär an der Hypophyse manifestieren, eher den PZNSL zuordnen. DLBCL, bei denen der hypophysäre Befall eine von mehreren systemischen Manifestationen darstellt, erscheinen hingegen eher den DLBCL, NOS zugehörig.
Ausblick
Anhand der vorliegenden morphologischen Beschreibungen und der immunhistochemischen Marker ist eine eindeutige Zuordnung aktuell nicht möglich. Diese Fragestellung kann wahrscheinlich auf genetischer Ebene mittels Gensequenzierung der nächsten Generation (NGS) und dem konsekutiven Abgleich mit bisher publizierten genetischen Datensätzen zum DLBCL, NOS und zum PZNSL geklärt werden. Dieses Vorhaben stellt eines der hämatologisch-onkologischen Forschungsprojekte am Bundeswehrkrankenhaus Ulm dar, welches in enger Kooperation mit der Hämatologie und Onkologie am UKSH Campus Lübeck und der Hämatopathologie Lübeck aktuell bearbeitet wird.
Fazit
Die vorliegende Arbeit unterstreicht die essenzielle Bedeutung einer klassifizierten onkologischen Expertise an den Bundeswehrkrankenhäusern. Durch das umfangreiche Repertoire an zur Verfügung stehender bildgebender und zudem histopathologischer und molekulargenetischer Diagnostik können Patienten mit komplizierten hämatologisch-onkologischen Erkrankungen adäquat im interdisziplinären Kontext versorgt werden. Da es sich bei diesen Erkrankungen um Systemerkrankungen handelt, trägt die Rekrutierung dieses Patientengutes maßgeblich zur vollumfassenden Ausbildung von Medizinern sämtlicher Disziplinen an den Bundeswehrkrankenhäusern bei. Der Schwerpunkt der onkologischen Forschung trägt darüber hinaus entscheidend zum näheren Verständnis von Schädigungen mit karzinogenem Potenzial durch die Exposition gegenüber elektromagnetischer und ionisierender Strahlung oder gegenüber Radionukliden bei.
Erklärungen
Die Verfasser erklären, dass kein Interessenkonflikt im Sinne der Richtlinien des International Committee of Medical Journal Editors besteht.
Die Verfasser möchten sich ausdrücklich für die Zusammenarbeit im Hinblick auf die Entstehung dieser Arbeit und die referenzpathologische Begutachtung bei Herrn Prof. Dr. med. A. Rosenwald, Instituts für Pathologie an der Universität Würzburg, bedanken.
Literatur
- Anila KR, Nair RA, Koshy SM, Jacob PM: Primary intravascular large B-cell lymphoma of pituitary. Indian J Pathol Microbiol 2012; 55(4): 549-551. mehr lesen
- Au WY, Kwong YL, Shek TW, Leung G, Ooi C: Diffuse large-cell B-cell lymphoma in a pituitary adenoma: an unusual cause of pituitary apoplexy. Am J Hematol 2000; 63(4): 231-232. mehr lesen
- Bayraktar S, Bassini W, Goodman M: Primary pituitary lymphoma: idiopathic anasarca with relapse in bone marrow only. Acta Haematol 2010; 123(2): 121-125. mehr lesen
- Bonnet C, Fillet G, Mounier N, Ganem G, Molina TJ, Thieblemont C, Ferme C, Quesnel B, Martin C, Gisselbrecht C et al: CHOP alone compared with CHOP plus radiotherapy for localized aggressive lymphoma in elderly patients: a study by the Groupe d'Etude des Lymphomes de l'Adulte. J Clin Oncol 2007; 25(7): 787-792. mehr lesen
- Carrasco CA, Rojas ZD, Chiorino R, Gonzalez G: Primary pituitary lymphoma in immunocompetent patient: diagnostic problems and prolonged follow-up. Pituitary 2012;15(1): 93-96. mehr lesen
- Chen SM, Chang CN, Wei KC, Jung SM, Chuang CC: Sellar lymphoma mimicking sphenoid infection presenting with cavernous sinus syndrome. J Clin Neurosci 2008; 15(10): 1148-1151. mehr lesen
- Ezzat S, Asa SL, Couldwell WT, Barr CE, Dodge WE, Vance ML, McCutcheon IE: The prevalence of pituitary adenomas: a systematic review. Cancer 2004; 101(3): 613-619. mehr lesen
- Ferreri AJ, Cwynarski K, Pulczynski E, Ponzoni M, Deckert M, Politi LS, Torri V, Fox CP, Rosee PL, Schorb E et al: Chemoimmunotherapy with methotrexate, cytarabine, thiotepa, and rituximab (MATRix regimen) in patients with primary CNS lymphoma: results of the first randomisation of the International Extranodal Lymphoma Study Group-32 (IELSG32) phase 2 trial. Lancet Haematol 2016; 3(5): e217-227. mehr lesen
- Freeman CL, Savage KJ, Villa D, Scott DW, Srour L, Gerrie AS, Brown MJ, Slack GW, Farinha P, Skinnider B: Long-Term Results of PET-Guided Radiation Therapy in Patients with Advanced-Stage Diffuse Large B-Cell Lymphoma Treated with R-CHOP in British Columbia. Blood 2017; 130 (Suppl. 1): 823. mehr lesen
- Grommes C, DeAngelis LM: Primary CNS Lymphoma. J Clin Oncol 2017; 35(21): 2410-2418. mehr lesen
- Guerrero-Perez F, Marengo AP, Vidal N, Iglesias P, Villabona C: Primary tumors of the posterior pituitary: A systematic review. Rev Endocr Metab Disord 2019; 20(2): 219-238. mehr lesen
- Hans CP, Weisenburger DD, Greiner TC, Gascoyne RD, Delabie J, Ott G, Muller-Hermelink HK, Campo E, Braziel RM, Jaffe ES et al: Confirmation of the molecular classification of diffuse large B-cell lymphoma by immunohistochemistry using a tissue microarray. Blood 2004; 103(1): 275-282. mehr lesen
- Hayasaka K, Koyama M, Yamashita T: Primary pituitary lymphoma diagnosis by FDG-PET/CT. Clin Nucl Med 2010; 35(3): 205. mehr lesen
- Heaney AP: Clinical review: Pituitary carcinoma: difficult diagnosis and treatment. J Clin Endocrinol Metab 2011; 96(12): 3649-3660. mehr lesen
- Horning SJ, Weller E, Kim K, Earle JD, O'Connell MJ, Habermann TM, Glick JH: Chemotherapy with or without radiotherapy in limited-stage diffuse aggressive non-Hodgkin's lymphoma: Eastern Cooperative Oncology Group study 1484. J Clin Oncol 2004; 22(15): 3032-3038. mehr lesen
- Horvat M, Zadnik V, Juznic Setina T, Boltezar L, Pahole Golicnik J, Novakovic S, Jezersek Novakovic B: Diffuse large B-cell lymphoma: 10 years' real-world clinical experience with rituximab plus cyclophosphamide, doxorubicin, vincristine and prednisolone. Oncol Lett 2018; 15(3): 3602-3609. mehr lesen
- Huang YY, Lin SF, Dunn P, Wai YY, Hsueh C, Tsai JS: Primary pituitary lymphoma presenting as hypophysitis. Endocr J 2005; 52(5): 543-549. mehr lesen
- Hussain S, Hallam S, Beltran L, Haroon A, Majumdar K, Shamash J, Sivapackianathan R, Drake W: Intravascular large B-cell lymphoma presenting as a pituitary mass with bilateral adrenal enlargement and haemophagocytic lymphohistiocytosis. Br J Haematol 2018; 181(6): 851-852. mehr lesen
- Katz BJ, Jones RE, Digre KB, Warner JE, Moore KR: Panhypopituitarism as an initial manifestation of primary central nervous system non-Hodgkin's lymphoma. Endocr Pract 2003; 9(4): 296-300. mehr lesen
- Kaufmann TJ, Lopes MB, Laws ER, Jr., Lipper MH: Primary sellar lymphoma: radiologic and pathologic findings in two patients. AJNR Am J Neuroradiol 2002; 23(3): 364-367. mehr lesen
- Kuhn D, Buchfelder M, Brabletz T, Paulus W: Intrasellar malignant lymphoma developing within pituitary adenoma. Acta Neuropathol 1999; 97(3): 311-316. mehr lesen
- Kumabe A, Kenzaka T, Nishimura Y, Aikawa M, Mori M, Matsumura M: A rare case of anasarca caused by infiltration of the pituitary gland by diffuse large B-cell lymphoma. BMC Endocr Disord 2015; 15: 10. mehr lesen
- Lake MG, Krook LS, Cruz SV: Pituitary adenomas: an overview. Am Fam Physician 2013; 88(5): 319-327. mehr lesen
- Landman RE, Wardlaw SL, McConnell RJ, Khandji AG, Bruce JN, Freda PU: Pituitary lymphoma presenting as fever of unknown origin. J Clin Endocrinol Metab 2001; 86(4): 1470-1476. mehr lesen
- Leon-Suarez A, Roldan-Sarmiento P, Gomez-Samano MA et al.: Infundibulo-hypophysitis-like radiological image in a patient with pituitary infiltration of a diffuse large B-cell non-Hodgkin lymphoma. Endocrinol Diabetes Metab Case Rep 2016; 1: 10.1530/EDM-16-0103. mehr lesen
- Li Y, Zhang Y, Xu J, Chen N: Primary pituitary lymphoma in an immunocompetent patient: a rare clinical entity. J Neurol 2012; 259(2): 297-305. mehr lesen
- Martinez JH, Davila Martinez M, Mercado de Gorgola M, Montalvo LF, Tome JE: The coexistence of an intrasellar adenoma, lymphocytic hypophysitis, and primary pituitary lymphoma in a patient with acromegaly. Case Rep Endocrinol 2011; 2011: 941738. mehr lesen
- Mathiasen RA, Jarrahy R, Cha ST, Kovacs K, Herman VS, Ginsberg E, Shahinian HK: Pituitary lymphoma: a case report and literature review. Pituitary 2000; 2(4): 283-287. mehr lesen
- Molitch ME: Diagnosis and Treatment of Pituitary Adenomas: A Review. JAMA 2017; 317(5): 516-524. mehr lesen
- Morita S, Ariyasu H, Ohiwa T, Akamizu T: Rapidly Progressing Pituitary Mass in B-cell Lymphoma. Intern Med 2019; 58(10): 1525-1526. mehr lesen
- Moshkin O, Muller P, Scheithauer BW, Juco J, Horvath E, Patterson BJ, Kamel-Reid S, Kovacs K: Primary pituitary lymphoma: a histological, immunohistochemical, and ultrastructural study with literature review. Endocr Pathol 2009; 20(1): 46-49. mehr lesen
- Nakashima Y, Shiratsuchi M, Abe I et al.: Pituitary and adrenal involvement in diffuse large B-cell lymphoma, with recovery of their function after chemotherapy. BMC Endocr Disord 2013; 13: 45. mehr lesen
- Papanastasiou L, Pappa T, Dasou A, Kyrodimou E, Kontogeorgos G, Samara C, Bacaracos P, Galanopoulos A, Piaditis G: Case report: Primary pituitary non-Hodgkin's lymphoma developed following surgery and radiation of a pituitary macroadenoma. Hormones (Athens) 2012; 11(4): 488-494. mehr lesen
- Rainsbury P, Mitchell-Innes A, Clifton N, Khalil H: Primary lymphoma of the pituitary gland: an unusual cause of hemianopia in an immunocompetent patient. JRSM Short Rep 2012; 3(8): 55. mehr lesen
- Ravindra VM, Raheja A, Corn H, Driscoll M, Welt C, Simmons DL, Couldwell WT: Primary pituitary diffuse large B-cell lymphoma with somatotroph hyperplasia and acromegaly: case report. J Neurosurg 2017; 126(5): 1725-1730. mehr lesen
- Rosenwald A, Bens S, Advani R, Barrans S, Copie-Bergman C, Elsensohn MH, Natkunam Y, Calaminici M, Sander B, Baia M et al: Prognostic Significance of MYC Rearrangement and Translocation Partner in Diffuse Large B-Cell Lymphoma: A Study by the Lunenburg Lymphoma Biomarker Consortium. J Clin Oncol 2019; 37(35): 3359-3368. mehr lesen
- Rudnik A, Larysz D, Blamek S, Larysz P, Bierzynska-Macyszyn G, Wlaszczuk P, Bazowski P: Primary pituitary lymphoma. Folia Neuropathol 2007; 45(3): 144-148. mehr lesen
- Samaratunga H, Perry-Keene D, Apel RL: Primary Lymphoma of Pituitary Gland: A Neoplasm of Acquired Malt? Endocr Pathol 1997; 8(4): 335-341. mehr lesen
- Schlegel U, Schmidt-Wolf IG, Deckert M: Primary CNS lymphoma: clinical presentation, pathological classification, molecular pathogenesis and treatment. J Neurol Sci 2000; 181(1-2): 1-12. mehr lesen
- Shaw JA, Strachan FM, Sawers HA, Bevan JS: Non-Hodgkin lymphoma with panhypopituitarism, hyperprolactinaemia and sixth nerve palsy. J R Soc Med 1997; 90(5): 274-275. mehr lesen
- Singh VP, Mahapatra AK, Dinde AK: Sellar-suprasellar primary malignant lymphoma: case report. Indian J Cancer 1993; 30(2): 88-91. mehr lesen
- Stegink JA, Shegal V, Konig M: A unique case of central hypopituitarism and central diabetes insipidus caused by diffuse large B cell lymphoma. AACE Clinical Case Reports 2019; 5(1): e22-e26. mehr lesen
- Stewart BW, Wild CP: World cancer report 2014. WHO IARC PublicationsPublic Health 2014.
- Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, Advani R, Ghielmini M, Salles GA, Zelenetz AD et al: The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016; 127(20): 2375-2390. mehr lesen
- Tamer G, Kartal I, Aral F: Pituitary infiltration by non-Hodgkin's lymphoma: a case report. J Med Case Rep 2009; 3: 9293. mehr lesen
- Tarabay A, Cossu G, Berhouma M, Levivier M, Daniel RT, Messerer M: Primary pituitary lymphoma: an update of the literature. J Neurooncol 2016; 130(3): 383-395. mehr lesen
- Wilkie MD, Al-Mahfoudh R, Javadpour M: A rare case of primary sellar lymphoma presenting a diagnostic challenge. Br J Neurosurg 2012; 26(5):782-783. mehr lesen
- Wolfe SQ HB, Barker J, Benveniste RJ: Primary central nervous system lymphoma mimicking pituitary apoplexy: case report. Pituitary 2009; 12(1): 76-79. mehr lesen
Manuskriptdaten
Eingereicht: 14. Februar 2020
Nach Überarbeitung angenommen: 21. März 2020
Zitierweise
Rother C, Gebauer N, Mayer T, Riecke A, Müller M, Hackenbroch C, Schulz C, Steinestel K, Witte HM: Das diffus großzellige B-Zell Lymphom der Hypophyse: Fallbericht einer seltenen Manifestation und Einordnung in die Literatur. WMM 2020; 64(5): 157-164.
Für die Verfasser
Stabsarzt Hanno M. Witte
Bundeswehrkrankenhaus Ulm
Klinik für Innere Medizin / Hämatologie und Onkologie
Oberer Eselsberg 40, 89081 Ulm
E-Mail: hanno.witte@uksh.de
Manuscript Data
Submitted: 14 February 2020
After revision accepted: 21 March 2020
Citation:
Rother C, Gebauer N, Mayer T, Riecke A, Müller M, Hackenbroch C, Schulz C, Steinestel K, Witte HM: Diffuse large B-cell lymphoma of the pituitary gland: Case report of a rare diesease and review of literature. WMM 2020; 64(5): 157-164.
For the authors
Captain (MC) Hanno M. Witte
Bundeswehr Hospital Ulm
Department for Internal Medicine / Hematology and Oncology
Oberer Eselsberg 40, D-89081 Ulm
E-Mail: hanno.witte@uksh.de
1Beiträge der Verfasser:
Studienkonzept: Rother, Witte; Datenerfassung: Rother, Riecke, Witte; Datenanalyse und Erstellung von Tabellen und Abbildungen: Steinestel, Gebauer, Hackenbroch, Rother, Witte; Erstfassung des Manuskripts: Rother, Witte. Korrektur des Manuskripts: Alle Autoren
2 IELSG32 = International Extranodal Lymphoma Study Group-32
3 CNS-IPI = Central Nervous System – International Prognostic Index: Score zur Abschätzung einer zentralen Manifestation für Patienten mit DLBCL